
Steric control of dioxygen activation pathways for MnII complexes supported by pentadentate, amide-containing ligands†

Abstract
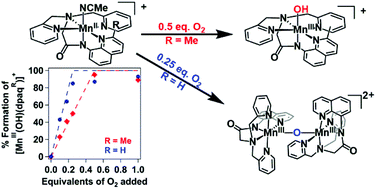
Dioxygen activation at manganese centers is well known in nature, but synthetic manganese systems capable of utilizing O2 as an oxidant are relatively uncommon. These present investigations probe the dioxygen activation pathways of two mononuclear MnII complexes supported by pentacoordinate amide-containing ligands, [MnII(dpaq)](OTf) and the sterically modified [MnII(dpaq2Me)](OTf). Dioxygen titration experiments demonstrate that [MnII(dpaq)](OTf) reacts with O2 to form [MnIII(OH)(dpaq)](OTf) according to a 4 : 1 Mn : O2 stoichiometry. This stoichiometry is consistent with a pathway involving comproportionation between a MnIV–oxo species and residual MnII complex to form a (μ-oxo)dimanganese(III,III) species that is hydrolyzed by water to give the MnIII–hydroxo product. In contrast, the sterically modified [MnII(dpaq2Me)](OTf) complex was found to react with O2 according to a 2 : 1 Mn : O2 stoichiometry. This stoichiometry is indicative of a pathway in which a MnIV–oxo intermediate abstracts a hydrogen atom from solvent instead of undergoing comproportionation with the MnII starting complex. Isotopic labeling experiments, in which the oxygenation of the MnII complexes was carried out in deuterated solvent, supported this change in pathway. The oxygenation of [MnII(dpaq)](OTf) did not result in any deuterium incorporation in the MnIII–hydroxo product, while the oxygenation of [MnII(dpaq2Me)](OTf) in d3-MeCN showed [MnIII(OD)(dpaq2Me)]+ formation. Taken together, these observations highlight the use of steric effects as a means to select which intermediates form along dioxygen activation pathways.
 Please wait while we load your content...
Please wait while we load your content...

