
Catalytic intramolecular hydroamination of aminoallenes using titanium complexes of chiral, tridentate, dianionic imine-diol ligands†

Abstract
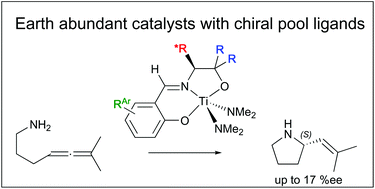
Alkylation of D- or L-phenylalanine or valine alkyl esters was carried out using methyl or phenyl Grignard reagents. Subsequent condensation with salicylaldehyde, 3,5-di-tert-butylsalicylaldehyde, or 5-fluorosalicylaldehyde formed tridentate, X2L type, Schiff base ligands. Chiral shift NMR confirmed retention of stereochemistry during synthesis. X-ray crystal structures of four of the ligands show either inter- or intramolecular hydrogen bonding interactions. The ligands coordinate to the titanium reagents Ti(NMe2)4 or TiCl(NMe2)3 by protonolysis and displacement of two equivalents of HNMe2. The crystal structure of one example of Ti(X2L)Cl(NMe2) was determined and the complex has a distorted square pyramidal geometry with an axial NMe2 ligand. The bis-dimethylamide complexes are active catalysts for the ring closing hydroamination of di- and trisubstituted aminoallenes. The reaction of hepta-4,5-dienylamine at 135 °C with 5 mol% catalyst gives a mixture of 6-ethyl-2,3,4,5-tetrahydropyridine (40–72%) and both Z- and E-2-propenyl-pyrrolidine (25–52%). The ring closing reaction of 6-methyl-hepta-4,5-dienylamine at 135 °C with 5 mol% catalyst gives exclusively 2-(2-methyl-propenyl)-pyrrolidine. The pyrrolidine products are obtained with enantiomeric excesses up to 17%.
- This article is part of the themed collection: The central role of the d-block metals in the periodic table
 Please wait while we load your content...
Please wait while we load your content...

