
Predicting aggregation energy for single atom bimetallic catalysts on clean and O* adsorbed surfaces through machine learning models†

Abstract
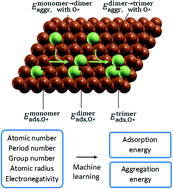
Single atom catalysts have received increased attention due to their unprecedented high metal dispersion. Recent progress in machine learning (ML) has significantly aided in the rational design of such heterogeneous catalysts, often dedicated to ML prediction of adsorption properties. A common shortfall, however, is the neglect of the thermodynamic stability of the assumed site and adsorbate-induced structural changes. Here, we developed ML models to predict the thermodynamic stability of a single-atom and related site of 38 different elements alloyed with Cu, trained with DFT calculations. To account for adsorbate-induced effects, we placed a monoatomic oxygen adsorbate in the vicinity of the dopant site. A Gaussian process regression (GPR) model performed best with a mean absolute error (MAE) of less than 0.08 eV for aggregation energy. The same performance was achieved with an even smaller training dataset using an active learning algorithm, producing a one-third time saving. Similarly, our ML prediction of O* adsorption energy gave a MAE of 0.08 eV. Furthermore, the ML model is extendable to different substrates (in addition to Cu), different adsorbates (in addition to O*), and larger cluster sizes (greater than trimer), demonstrating the potential of addressing large number of degrees of freedom and orders of magnitude time saving.
 Please wait while we load your content...
Please wait while we load your content...

