
Theoretical investigation on the hydrogen evolution reaction mechanism at MoS2 heterostructures: the essential role of the 1T/2H phase interface†

Abstract
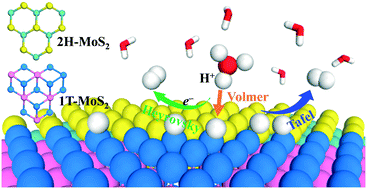
MoS2 with mixed 1T and 2H phases has exhibited excellent catalytic activity for the hydrogen evolution reaction (HER) in recent experiments. However the essential role played by the 1T/2H phase interface is still obscure. Herein, periodic density functional theory (DFT) calculations have been performed to study the HER mechanism at two types of 1T/2H phase interfaces of MoS2 (“zigzag” and “armchair”). By analyzing the free energy of atomic hydrogen adsorption (ΔGH) as the descriptor, we suggest that the optimum evolution of H2 proceeds at ∼10% H coverage for the “zigzag” and “armchair” interfaces. Under this H coverage, the Volmer–Tafel mechanism is the dominant reaction pathway for both interfaces, in which the Volmer reaction is the rate determining step, and the HER would proceed more easily at the “zigzag” interface with lower energy barriers. The results show that the HER activity along the 1T/2H phase interface is comparable with those at the Mo-edge of 2H MoS2 and the basal plane of 1T MoS2. In addition, we investigate the effect of metal (Fe, Co, Ni, and Zn) and non-metal (N, P, and O) dopants for the “zigzag” type interface, and propose that the HER activity could be improved by doping with Ni for the interfacial Mo atom or with N for the interfacial S atom.
 Please wait while we load your content...
Please wait while we load your content...

