
Reactivity trends of the MoVOx mixed metal oxide catalyst from density functional modeling†

Abstract
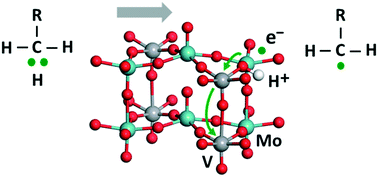
We modeled the oxidative dehydrogenation of hydrocarbons at various oxygen sites in the basal plane of the M1 (orthorhombic) phase of the MoVOx catalyst material, determining hydrogen adsorption energies by applying a hybrid DFT method to three-layer slab models. H adsorption is favored at bridging O centers over terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites. Thereby, the unpaired electron, originating from the H atom, was calculated to localize preferentially at V centers, except if there are no V5+ centers near the H adsorption site; in the latter case reduction of a Mo6+ center is predicted. A reducing electron may transfer from a metal center in the surface layer to a sub-surface V center, with an associated stabilization of ∼10–90 kJ mol−1. We also probed the initial step for hydrolyzing the MoVOx surface. The adsorption of a water molecule is calculated to be more favorable, by 30 kJ mol−1, at a surface Mo6+ center than at a surface V5+ center (by 22 kJ mol−1 over a surface V4+ center). Remarkably, the trends in the reaction free energies of hydrogenation and hydrolysis are quite similar for various oxygen sites and the position of the newly created polaron in the surface model. The specific energetics, however, depends notably on the latter factors.
O sites. Thereby, the unpaired electron, originating from the H atom, was calculated to localize preferentially at V centers, except if there are no V5+ centers near the H adsorption site; in the latter case reduction of a Mo6+ center is predicted. A reducing electron may transfer from a metal center in the surface layer to a sub-surface V center, with an associated stabilization of ∼10–90 kJ mol−1. We also probed the initial step for hydrolyzing the MoVOx surface. The adsorption of a water molecule is calculated to be more favorable, by 30 kJ mol−1, at a surface Mo6+ center than at a surface V5+ center (by 22 kJ mol−1 over a surface V4+ center). Remarkably, the trends in the reaction free energies of hydrogenation and hydrolysis are quite similar for various oxygen sites and the position of the newly created polaron in the surface model. The specific energetics, however, depends notably on the latter factors.
 Please wait while we load your content...
Please wait while we load your content...

