
Relatives of cyanomethylene: replacement of the divalent carbon by B−, N+, Al−, Si, P+, Ga−, Ge, and As+†

Abstract
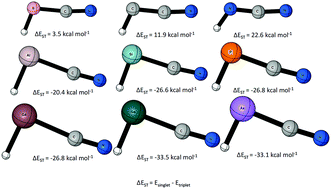
The lowest lying singlet and triplet states of HBCN−, HCCN, HNCN+, HAlCN−, HSiCN, HPCN+, HGaCN−, HGeCN, and HAsCN+ were studied using the CCSDT(Q)/CBS//CCSD(T)/aug-cc-pVQZ level of theory. Periodic trends in geometries, singlet–triplet gaps, and barriers to linearity were established and analyzed. The first row increasingly favors the triplet state, with a singlet–triplet gap (ΔEST = Esinglet − Etriplet) of 3.5 kcal mol−1, 11.9 kcal mol−1, and 22.6 kcal mol−1, respectively, for HBCN−, HCCN, and HNCN+. The second row increasing favors the singlet state, with singlet–triplet gaps of −20.4 kcal mol−1 (HAlCN−), −26.6 kcal mol−1 (HSiCN), and −26.8 kcal mol−1 (HPCN+). The third row also favors the singlet state, with singlet–triplet gaps of −26.8 kcal mol−1 (HGaCN−), −33.5 kcal mol−1 (HGeCN), and −33.1 kcal mol−1 (HAsCN+). The HXCN species have larger absolute singlet–triplet energy gaps compared to their parent species XH2 except for the case of X = N+. The effect of the substitution of hydrogen with a cyano group was analyzed with isodesmic bond separation analysis and NBO.
 Please wait while we load your content...
Please wait while we load your content...

