
Electron transfer in extended systems: characterization by periodic density functional theory including the electronic coupling†
 a
and
Michel
Dupuis
*a
a
and
Michel
Dupuis
*a
Abstract
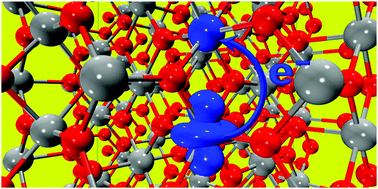
We describe a new computer implementation of electron transfer (ET) theory in extended systems treated by periodic density functional theory (DFT), including the calculation of the electronic coupling transition element VAB. In particular, the development opens up the full characterization of electron transfer in the solid state. The approach is valid for any single-determinant wavefunction with localized character representing the electronic structure of the system, from Hartree–Fock (HF) theory, to density functional theory (DFT), hybrid DFT theory, DFT+U theory, and constrained DFT (cDFT) theory. The implementation in CP2K reuses the high-performance functions of the code. The computational cost is equivalent to only one iteration of an HF calculation. We present test calculations for electron transfer in a number of systems, including a 1D-model of ferric oxide, hematite Fe2O3, rutile TiO2, and finally bismuth vanadate BiVO4.
- This article is part of the themed collection: Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces
 Please wait while we load your content...
Please wait while we load your content...

