
Site dependent reactivity of Pt single atoms on anatase TiO2(101) in an aqueous environment†
 ab
Annabella
Selloni
*b
and
ab
Annabella
Selloni
*b
and
Abstract
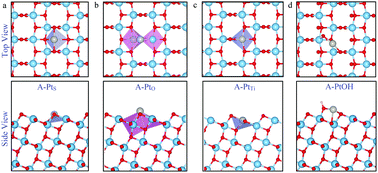
The TiO2–Pt–water interface is of great relevance in photocatalysis where Pt is widely used as a co-catalyst for enhancing hydrogen evolution in aqueous TiO2. Using ab initio molecular dynamics, we investigated this interface focusing on Pt single atoms supported on anatase TiO2(101) in a water environment. Based on recent experiments showing a broad distribution of Pt coordination sites in TiO2, we examined six distinct single-Pt supported species with different nominal Pt oxidation states, namely: Pt, PtOH, and PtO2 species adsorbed on the stoichiometric surface; Pt adsorbed at a surface oxygen vacancy (Ov); and Pt substituting a surface Ti cation (PtTi), both without and with an accompanying Ov (PtTi + Ov). As found for the pristine anatase surface, interfacial water remained intact in the presence of a nearly neutral Pt adatom within the time duration of our simulations (∼15 ps). Similarly, no (or only temporary) water dissociation was observed at the PtTi + Ov and PtO2 interfaces, due to the formation of very stable planar Pt coordination structures that interact only weakly with water. In contrast, water dissociated with OH− (H+) on the Pt atom when this substituted a surface Ti (oxygen) ion as well as on PtOH. The significant proton affinity of Pt atoms at surface oxygen vacancies suggests that negatively charged Pt species are particularly efficient at catalyzing hydrogen evolution in aqueous TiO2.
- This article is part of the themed collections: Frontiers in Molecular Simulation of Solvated Ions, Molecules and Interfaces and 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

