
Theoretical investigation of the cation antisite defect in layer-structured cathode materials for Li-ion batteries

Abstract
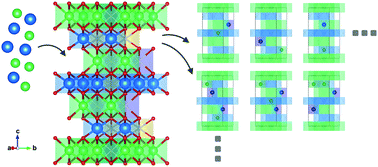
The formation of cation antisite defects in layer-structured LiTMO2 cathode materials for Li-ion batteries is investigated, where TM represents Ni or Co. Energetically possible arrangements of cation antisite defects are searched by a bottom-up method in which cations are added to a frame supercell that contains some vacant cation lattice points, using a particle swarm optimization algorithm combined with a density functional theory method. The equilibrium concentration of antisite defects is quantified through a calculation model in which an ensemble crystal system is assembled by statistically combining the constructed supercell units. The behavior of antisite defect formation in LiNiO2 and LiCoO2 is compared, and the effect of Co or Mn introduction to LiNiO2 is examined. The underlying mechanism is investigated in terms of the covalent and ionic bonding characteristics, TM oxidation state, and structural changes in the cation sites, based on analyses of the crystal orbital Hamilton population, partial charge, and bond lengths. It is concluded that the structural distortion of the cation sites and the superexchange interaction involved with cation mixing are the two main factors determining the concentration of cation antisite defects in layer-structured cathode materials.
 Please wait while we load your content...
Please wait while we load your content...

