
p-Type conductivity mechanism and defect structure of nitrogen-doped LiNbO3 from first-principles calculations†
 b
Hongde
Liu,
*a
b
Hongde
Liu,
*a
Abstract
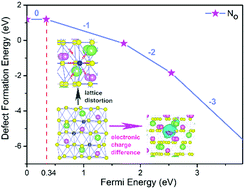
Most metal-doped lithium niobates (LiNbO3, LN) exhibit n-type conductivity. The absence of p-type conductive LiNbO3 limits its application. Based on the finding that p-type conductive LiNbO3 can be realized by doping with a non-metallic element N, we investigate the most stable defect configurations and formation energies of LiNbO3 doped with non-metal nitrogen (LN:N) by first-principles calculations. Nitrogen substitution, interstitial and quasi-substitution point defects in different sites and their effects were explored. The results show that N prefers to occupy the oxygen site with only little lattice distortion. Ab initio molecular dynamics (AIMD) simulations confirm the structural stability of an N ion occupying the O site. The charge-state transition level ε(0/−1) slightly above the valence band maximum (VBM) indicates that N point defects would contribute to p-type conductivity of LiNbO3. The analysis of the band structure reveals that the partially filled impurity levels can accommodate electrons that jump from valence bands and result in holes to become the main charge carriers. The calculation not only explains the occurrence of p-type conductivity in LN:N but also provides a simple and efficient way to discover p-type conductive candidates in numerous doped LiNbO3 crystals.
 Please wait while we load your content...
Please wait while we load your content...

