
Computational study of aromaticity, 1H NMR spectra and intermolecular interactions of twisted thia-norhexaphyrin and its multiply annulated polypyrrolic derivatives†

Abstract
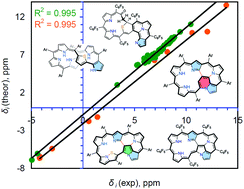
The recently synthesized twisted thia-norhexaphyrin and its multiply annulated polypyrrolic derivatives have been studied computationally. Gauge-including magnetically induced current calculations predict a global nonaromatic character of the initial thia-norhexaphyrin due to the highly-twisted conformation of the macrocycle. Upon the oxidation of the thia-norhexaphyrin four multiply annulated polypyrrolic aromatic macrocycles are formed for which the global aromatic character is confirmed in agreement with experimentally measured 1H NMR spectra. The calculation of the proton chemical shifts for the studied compounds by direct comparison with the tetramethylsilane standard leads to a significant mean absolute error. At the same time a linear regression procedure for the two selected groups of protons (CH and NH protons) provides much better values of calculated chemical shifts and tight correlation with experiment. The separate consideration of NH protons is motivated by the numerous intermolecular hydrogen bonds in which the protons are involved, which induce considerable upfield shifts, leading to a significant underestimation of the corresponding chemical shifts. Such a selected correlation can be used for accurate estimation of proton chemical shifts of the related porphyrinoids. Bader's theory of Atoms in Molecules has been applied for the studied twisted thia-norhexaphyrin and its multiply annulated polypyrrolic derivatives to characterize intramolecular H-bonds and other non-covalent interactions.
 Please wait while we load your content...
Please wait while we load your content...

