
How to calculate charge mobility in molecular materials from surface hopping non-adiabatic molecular dynamics – beyond the hopping/band paradigm

Abstract
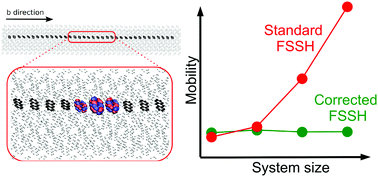
Charge transport in high mobility organic semiconductors is in an intermediate regime between small polaron hopping and band transport limits. We have recently shown that surface hopping non-adiabatic molecular dynamics is a powerful method for prediction of charge transport mechanisms in organic materials and for near-quantitative prediction of charge mobilities at room temperature where the effects of nuclear zero-point motion and tunneling are still relatively small [S. Giannini et al., Nat. Commun., 2019, 10, 3843]. Here we assess and critically discuss the extensions to Tully's original method that have led to this success: (i) correction for missing electronic decoherence, (ii) detection of trivial crossings and (iii) removal of decoherence correction-induced spurious charge transfer. If any one of these corrections is not included, the charge mobility diverges with system size, each for different physical reasons. Yet if they are included, convergence with system size, detailed balance and good internal consistency are achieved.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

