
Comparing the performances of various density functionals for modelling the mechanisms and kinetics of bimolecular free radical reactions in aqueous solution†
 a
and
Ivan
Ljubić
*a
a
and
Ivan
Ljubić
*a
Abstract
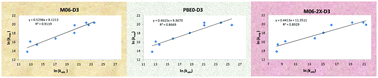
We carried out an investigation of the performances of 18 density functionals (DFs) for modelling the mechanisms and kinetics of the aqueous phase reactions between the α-hydroxyisopropyl radical and 9 organic substrates. The primary goal was to evaluate the applicability of density functional theory specifically in conjunction with the polarizable continuum model (DFT/PCM) for a fully implicit description of the aqueous environment. Accordingly, the solute is augmented with the explicit molecule(s) of the water solvent only when it is confirmed that the water participates in the reaction mechanism directly and not just as a potential donor or acceptor of additional hydrogen bonds. The tested DFs are chosen by systematically ascending the Jacob's ladder of DFs with particular emphasis on the versatile Minnesota family. For most of the DFs we used the empirical corrections for the dispersion in accordance with Grimme's D3 or D3-BJ models. The optimum DFs are determined on the basis of the lowest mean absolute errors (MAEs) and the largest Pearson correlation coefficients (PCCs) with respect to the set of experimentally determined rate constants. The studied substrates are carbon tetrachloride (CCl4), chloroform (CHCl3), trichloroacetate (Cl3Ac−), chloral hydrate (ClH), iodoacetate (IAc−), iodoacetamide (IAm), 5-bromouracil (BrU), 5-nitrouracil (NO2U), and cysteamine (Cys+). The mechanisms that contribute dominantly to the observed rate constants are: chlorine abstractions for CCl4, CHCl3, Cl3Ac−, and ClH; proton-coupled electron transfer (PCET) for IAc−; water-assisted PCET and iodine abstraction for IAm; ortho-addition for BrU and NO2U; and hydrogen atom abstraction from the sulphur atom for Cys+. It is found that in the DFT/PCM setting climbing up the Jacob's ladder does not necessarily imply a systematically increasing accuracy. Thus, M06-D3 and PBE0-D3 exhibit the best performance according to the lowest MAEs (1.10 and 1.26 kcal mol−1 MAEs in the Gibbs free energies of activation), and M06-D3, M06-2X-D3 and MN15 according to the largest PCCs (0.95, 0.94, and 0.94). In a surprising contrast, the three tested double-hybrid DFs, B2PLYP-D3, DSD-PBEP86, and PBEQIDH, all exhibit comparatively large MAEs and poor PCCs, and therefore do not appear well-suited for use in the DFT/PCM framework.
 Please wait while we load your content...
Please wait while we load your content...

