
A computational study of hydrogen doping induced metal-to-insulator transition in CaFeO3, SrFeO3, BaFeO3 and SmMnO3†
 *a
*a
Abstract
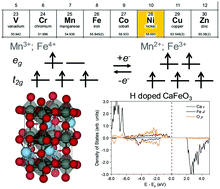
The metal-to-insulator transition (MIT) in rare earth perovskite oxides has drawn significant research interest for decades to unveil the underlying physics and develop novel electronic materials. Recently, chemical doping induced MIT in SmNiO3 has been observed experimentally, with its resistivity changed by eight orders of magnitude. The mechanism of switching from one singly occupied Ni eg orbital to two singly occupied eg orbitals upon doping has been proposed by experimentalists and verified by computation. Here, we tested if this mechanism can be generally applied to other perovskite oxides with non-Ni B site elements. We applied first principles density functional theory (DFT) to study a series of perovskite oxides, CaFeO3, SrFeO3, BaFeO3 and SmMnO3. We investigated the geometry and electronic structures of pure and hydrogen doped oxides. We found that pure CaFeO3, SrFeO3 and BaFeO3 are metallic while pure SmMnO3 has a small band gap of 0.69 eV. Upon hydrogen doping, band gap opening was predicted for all four oxides: HSE06 predicted band gap values of 1.58 eV, 1.40 eV, 1.20 eV and 2.55 eV for H-doped CaFeO3, SrFeO3, BaFeO3 and SmMnO3, respectively. This finding opens up research opportunities for exploring a broader range of materials for MIT to be used in optical and electronic devices.
 Please wait while we load your content...
Please wait while we load your content...

