
Accurate K-edge X-ray photoelectron and absorption spectra of g-C3N4 nanosheets by first-principles simulations and reinterpretations†

Abstract
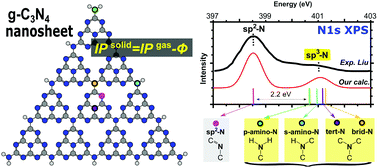
We performed a density functional theory (DFT) study on X-ray photoelectron (XPS) and absorption (XAS) spectra of graphitic carbon nitride (g-C3N4) nanosheets at the N and C K-edges. A combined cluster-periodic approach was employed to calculate XPS spectra, in which the core ionic potential (IP) of the solid 2D material was computed by subtracting the work function (obtained with periodic conditions) from the gas phase IP (obtained with large cluster models). With amino-terminated supermolecules of different sizes, we obtained convergent spectra and provide new assignments for 5 nitrogen [1 sp2; 4 sp3 (bridging, tertiary, and primary/secondary amino nitrogens)] and 4 carbon (all bonded with three nitrogens) local structures. A good agreement with experiments was obtained, with the N1s (C1s) main peak position differing by 0.1–0.2 eV (0.5–0.8 eV). Our simulations show that N1s XPS of pure g-C3N4 contains only two major features at 398.6 and 401.2 eV, contributed from sp2-N and sp3-N, respectively. The chemical shifts of all sp3-N are so close (deviating by 0.3–0.6 eV) that terminal amino groups –NHx (x = 1, 2) will only be distinguished in high-resolution measurements. In C1s XPS, all carbons show similar (deviation < 0.2 eV) IPs, as determined by the same nearest neighbors. We further excluded the effect of shake-up satellites that may change our XPS interpretations by equivalent core hole time-dependent DFT (ECH-TDDFT) simulations. The effect of vibronic coupling is small (redistribution is only 0.1–0.3 eV to the higher-energy region) in the N1s edge as estimated from the asymmetric main peak shape, and negligible in the C1s edge. Quicker size convergence was found in XAS than XPS. In N1s XAS, we identified a weak π* spectral feature at 400–401 eV for both –NHx and tertiary nitrogens. Our study provides a clear theoretical reference for X-ray spectral fingerprints of different local structures, which is useful for analysis of g-C3N4 based materials with various designed or unavoidable structural modifications. We also highlight our combined cluster-periodic approach in calculating the K-edge XPS spectra of general 2D materials which predicts accurate absolute values.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

