
Revised values for the X23 benchmark set of molecular crystals†

Abstract
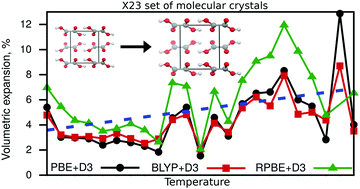
We present revised reference values for cell volumes and lattice energies for the widely used X23 benchmark set of molecular crystals by including the effect of thermal expansion. For this purpose, thermally-expanded structures were calculated via the quasi-harmonic approximation utilizing three dispersion-inclusive density-functional approximations. Experimental unit-cell volumes were back-corrected for thermal and zero-point energy effects, allowing now a direct comparison with lattice relaxations based on electronic energies. For the derivation of reference lattice energies, we utilized harmonic vibrational contributions averaged over four density-functional approximations. In addition, the new reference values also take the change in electronic and vibrational energy due to thermal expansion into account. This is accomplished by either utilizing experimentally determined cell volumes and heat capacities, or by relying on the quasi-harmonic approximation. The new X23b reference values obtained this way will enable a more accurate benchmark for the performance of computational methods for molecular crystals.
- This article is part of the themed collection: Bunsentagung 2020: Understanding Dispersion Interactions in Molecular Chemistry
 Please wait while we load your content...
Please wait while we load your content...

