
CASPT2, CASSCF and non-adiabatic molecular dynamics (NAMD) studies on the low-lying electronic states of 1H-1,2,3-triazole photolysis†

Abstract
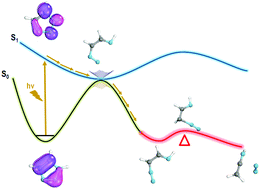
The photolysis mechanisms of 1H-1,2,3-triazole and 1H-1,2,3-benzotriazole were elucidated by employing multiconfigurational methods (CASSCF and CASPT2). The potential energy curves and crossing points for the low-lying excited states were analyzed. In addition to the static electronic structure calculations, non-adiabatic molecular dynamics (NAMD) was propagated at the CASSCF level using SHARC (Surface Hopping including ARbitrary Couplings) dynamics in order to verify the proposed static picture, thereby understanding the possible reaction paths and the time scale of the photo-induced events. The S1 state for 1H-1,2,3-triazole reached a conical intersection between the S0 and S1 surfaces on a time scale of 100 fs. The emerging picture of the reaction presented here is the rupture of the triazole ring in the S1 state and the relaxation through a conical intersection to the S0 state. On the S0 surface, the triazoles easily extrude N2 and then undergo the hetero-Wolff rearrangement forming ethanimine.
 Please wait while we load your content...
Please wait while we load your content...

