
Structure–thermodynamics relationship of schoepite from first-principles†
 *a
Eunja
Kim
b
*a
Eunja
Kim
b
Abstract
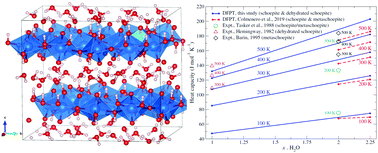
The relationship between the structure and thermodynamic properties of schoepite, an important uranyl phase with formula [(UO2)8O2(OH)12]·12H2O formed upon corrosion of UO2, has been investigated within the framework of density functional perturbation theory (DFPT). Experimental crystallographic lattice parameters are well reproduced in this study using standard DFT. Phonon calculations within the quasi-harmonic approximation predict standard molar entropy and isobaric heat capacity of S0 = 179.60 J mol−1 K−1 and C0P = 157.4 J mol−1 K−1 at 298.15 K, i.e., ∼6% and ∼4% larger than existing DFPT-D2 calculations. The computed variation of the standard molar isobaric heat capacity with water content from schoepite (UO3·xH2O, x = 2.25) to dehydrated schoepite (x = 1) is predicted to be essentially linear along isotherms ranging from 100 to 500 K. These findings have important implications for the dehydration of layered uranyl corrosion phases and hygroscopic materials.
 Please wait while we load your content...
Please wait while we load your content...

