
On principal features of organic electrolyte molecules in lithium ion battery performance†
 *a
and
Yoshitaka
Tateyama
bc
*a
and
Yoshitaka
Tateyama
bc
Abstract
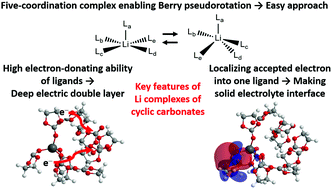
The lithium (Li) complexes of organic electrolyte solvents are theoretically investigated using the long-range correction for density functional theory in order to figure out the cause for the high performance of cyclic carbonate electrolytes in lithium ion batteries (LIBs). Calculations of the Li complexes with ethylene carbonate solvent molecules prove that ten ligand molecules should be incorporated to obtain near-degenerate four- and five-coordination optimum structures and dramatically improved orbital energies. The geometry optimizations of the Li complexes with thirteen types of organic solvent molecules give four-coordination neutral and five-coordination cation complexes for many solvent molecules. The five-coordination Li complexes are considered to use Berry pseudorotation to approach the electrodes from the Li atom. The calculated Koopmans, vertical and adiabatic ionization potentials and electron affinities show that near-degeneracy and structural deformation effects play significant roles in the electronic states of the Li complexes. Mulliken charge and dipole moment analyses indicate that the Li complexes of cyclic carbonates construct a deep electric double layer near electrodes due to the electron-donating ability of the ligand molecules. Molecular orbital analyses also explain that the Li complexes of cyclic carbonates easily construct a solid electrolyte interface, which contributes to Li ion conductance, by localizing the accepted electron to one ligand molecule. In conclusion, the Li complexes of cyclic carbonates have three main features: preference of five-coordination structures, high electron-donating ability of ligand molecules, and localization of the accepted electron to one ligand molecule.
 Please wait while we load your content...
Please wait while we load your content...

