
Manganese doping mechanism in a CsPbI2Br photovoltaic material: a first-principles study†

Abstract
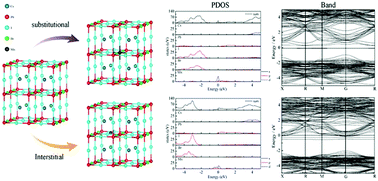
As a light absorbing material of perovskite solar cells, Mn-doped CsPbI2Br has a better phase stability than the undoped one. In order to deeply understand the doping mechanism of Mn, the effect of substitutional and interstitial Mn doping on the structural, electronic and optical properties of CsPbI2Br has been investigated by first-principles calculations based on density functional theory. It is found that the binding energy of both the substitutional and the interstitial Mn-doped CsPbI2Br is negative and the binding energy difference between them is only 2.8 meV, which indicates that both the substitutional and the interstitial doping structures should be stable for Mn-doped CsPbI2Br and the latter is slightly preferred over the former due to the lower binding energy. The lattice parameters of CsPbI2Br change oppositely for two Mn-doping cases. Based on the comparative analysis of the electronic structures for CsPbI2Br and Mn-doped CsPbI2Br, we found that the substitutional doping of Mn introduces intermediate bands near the Fermi level, making CsPbI2Br an intermediate band semiconductor; for the interstitial Mn-doped CsPbI2Br the Fermi level enters conduction bands, making it an n-type semiconductor material with enhanced conductivity. The complex dielectric function and the absorption spectrum of Mn-doped and undoped CsPbI2Br were calculated and are basically consistent with the experimental results.
 Please wait while we load your content...
Please wait while we load your content...

