
Size induced structural changes in maricite-NaFePO4: an in-depth study by experiment and simulations†
 c
and
Sevi
Murugavel
*a
c
and
Sevi
Murugavel
*a
Abstract
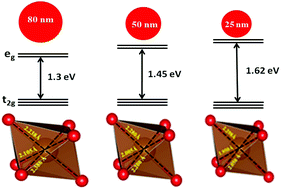
Rechargeable batteries based on the most abundant elements, such as sodium and iron, have a great potential in the development of cost effective sodium ion batteries for large scale energy storage devices. We report, for the first time, crystallite size dependent structural investigations on maricite-NaFePO4 through X-ray diffraction, X-ray absorption spectroscopy and theoretical simulations. Rietveld refinement analysis on the X-ray diffraction data reveals that a decrease in the unit cell parameters leads to volume contraction upon reduction in the crystallite size. Further, the atomic multiplet simulations on X-ray absorption spectra provide unequivocally a change in the site symmetry of transition metal ions. The high resolution oxygen K-edge spectra reveal a substantial change in the bonding character with the reduction of crystallite size, which is the fundamental cause for the change in the unit cell parameters of maricite-NaFePO4. In parallel, we performed first-principles density functional theory (DFT) calculations on maricite-NaFePO4 with different sodium ion vacancy concentrations. The obtained structural parameters are in excellent agreement with the experimental observations on the mesostructured maricite-NaFePO4. The volumetric changes with respect to crystallite size are related to the compressive strain, resulting in the improvement in the electronic diffusivity. The nano-crystalline maricite-NaFePO4 with improved kinetics will open a new avenue for its usage as a cathode material in sodium ion batteries.
 Please wait while we load your content...
Please wait while we load your content...

