
New insights into the origin of unstable sodium graphite intercalation compounds†
 c
and
Doreen
Mollenhauer
*ab
c
and
Doreen
Mollenhauer
*ab
Abstract
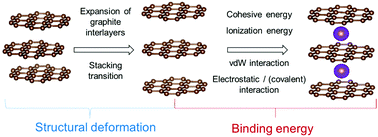
The thermodynamically unstable binary graphite intercalation compounds (GICs) with Na remain a main drawback preventing the implementation of Na-ion batteries in the market. In order to shed some light on the origin of Na–GICs instability, we investigate the structure and the energetics of different alkali metal (AM)–GICs by means of density functional theory (DFT) calculations with dispersion correction. We carefully consider different stages of AM–GICs for various AM concentrations and compare the results for Li, Na and K intercalation into graphite. In order to understand the compound stability, we investigated the interplay between the binding energy and the structural deformation due to the presence of AMs in graphite. Whereas the structural deformation energy linearly increases with the size of alkali metal ions, the binding energy passes through a maximum for Na–GIC. The analysis of different contributions to the binding energy allows to conclude that the alkali metal trend is broken for Li–GICs, not for Na–GICs. The high capacity for Li–GIC is a result of the small ion size of lithium. In addition to the mainly ionic binding nature, it allows to form a covalent contribution between lithium and graphite by orbital overlapping. In contrast, Na–GIC and K–GIC exhibit very small or hardly any covalent contribution. Furthermore, due to the small size of lithium the structural deformation energy cost also is small and allows van der Waals interactions between the graphite layers, which further enhance the stability of Li–GICs. For Na– and K–GICs, a higher energy amount for a structural deformation is needed and the stabilizing van der Waals interaction of graphite layers is weaker or hardly present.
 Please wait while we load your content...
Please wait while we load your content...

