
MARTINI-based simulation method for step-growth polymerization and its analysis by size exclusion characterization: a case study of cross-linked polyurethane†
 *a
*a
Abstract
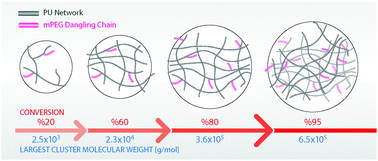
Simulation studies of step-growth polymerization, e.g., polymerization of polyurethane systems, hold great promise due to having complete control over the reaction conditions and being able to perform an in-depth analysis of network structures. In this work, we developed a (completely automated) simulation method based on a coarse-grained (CG) methodology, i.e., the MARTINI model, to study the cross-linking reaction of a diol, a tri-isocyanate molecule and one-hydroxyl functional molecule to form a polyurethane network without and with dangling chains. This method is capable of simulating the cross-linking reactions not only up to very high conversions, but also under rather complicated reaction conditions, i.e., a non-stoichiometric ratio of the reactants, solvent evaporation and multi-step addition of the reactants. We introduced a novel network analysis, similar to size-exclusion chromatography based on graph theory, to study the growth of the network during the polymerization process. By combining the reaction simulations with these analysis methods, a set of correlations between the reaction conditions, reaction mechanisms and final network structure and properties is revealed. For instance, a two-step addition of materials for the reaction, i.e., first the dangling chain to the tri-isocyanate and then the diol, leads to the highest integrated network structure. We observed that different reaction conditions lead to different glass transition temperatures (Tg) of the network due to the distinct differences in the final network structures obtained. For example, by addition of dangling chains to the network, the Tg decreases as compared to the network without dangling chains, as also is commonly observed experimentally.
 Please wait while we load your content...
Please wait while we load your content...

