
Spectroscopy of YO from first principles†

Abstract
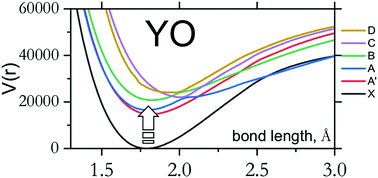
We report an ab initio study on the spectroscopy of the open-shell diatomic molecule yttrium oxide, YO. The study considers the six lowest doublet states, X2Σ+, A′2Δ, A2Π, B2Σ+, C2Π, D2Σ+, and a few higher-lying quartet states using high levels of electronic structure theory and accurate nuclear motion calculations. The coupled cluster singles, doubles, and perturbative triples, CCSD(T), and multireference configuration interaction (MRCI) methods are employed in conjunction with a relativistic pseudopotential on the yttrium atom and a series of correlation-consistent basis sets ranging in size from triple-ζ to quintuple-ζ quality. Core–valence correlation effects are taken into account and complete basis set limit extrapolation is performed for CCSD(T). Spin–orbit coupling is included through the use of both MRCI state-interaction with spin–orbit (SI-SO) approach and four-component relativistic equation-of-motion CCSD calculations. Using the ab initio data for bond lengths ranging from 1.0 to 2.5 Å, we compute 6 potential energy, 12 spin–orbit, 8 electronic angular momentum, 6 electric dipole moment and 12 transition dipole moment (4 parallel and 8 perpendicular) curves which provide a complete description of the spectroscopy of the system of six lowest doublet states. The Duo nuclear motion program is used to solve the coupled nuclear motion Schrödinger equation for these six electronic states. The spectra of 89Y16O simulated for different temperatures are compared with several available high resolution experimental studies; good agreement is found once minor adjustments are made to the electronic excitation energies.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

