
Optical spectra of molecular aggregates and crystals: testing approximation schemes†
 a
and
A.
Painelli
*a
a
and
A.
Painelli
*a
Abstract
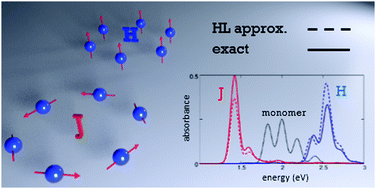
The interplay between exciton delocalization and molecular vibrations profoundly affects optical spectra of molecular aggregates and crystals. The exciton motion occurs on a similar timescale as molecular vibrations, leading to a complex and intrinsically non-adiabatic problem that has been handled over the years introducing several approximation schemes. Here we discuss systems where intermolecular distances are large enough so that only electrostatic intermolecular interactions enter into play and can be treated in the dipolar approximation. Moreover, we only account for interactions between transition dipole moments, as relevant to symmetric molecules, with negligible permanent (multi)polar moments in the ground and low-lying excited states. Translational symmetry is fully exploited to obtain numerically exact solutions of the relevant Hamiltonian for systems of comparatively large size. This offers a unique opportunity to assess the reliability of different approximation schemes. The so-called Heitler–London approximation, only accounting for the effects of intermolecular interactions among degenerate electronic states, leads to the celebrated exciton model, widely adopted to describe optical spectra of molecular aggregates and crystals. We demonstrate that, mainly due to a cancellation of errors, the exciton model approximates well the position of exciton bands and reasonably well the bandshapes, but it fails to predict spectral intensities, leading to underestimated intensities in J-aggregates and overestimated intensities in H-aggregates. This general result is validated against an exact sum-rule. Finally, we address the validity of several approximation schemes adopted to reduce the dimension of the vibrational basis.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

