
Cohesive properties of the crystalline phases of twenty proteinogenic α-aminoacids from first-principles calculations†
 *a
and
Michal
Fulem
a
*a
and
Michal
Fulem
a
Abstract
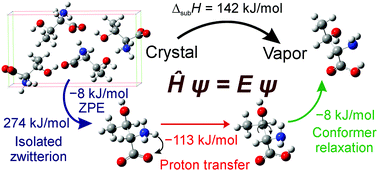
Cohesive properties (lattice and cohesive energy of the crystal and corresponding sublimation enthalpy) of the complete set of twenty enantiopure anhydrous proteinogenic amino acids are investigated using first-principles calculations. In contrast to neutral amino acid molecules in the vapor phase, all amino acids form crystals in their zwitterionic form. Therefore, reliable ab initio calculations of the proton transfer energy are an indispensable step of such calculations. Simplifying procedures, designed to rationalize the computational cost of the quasi-harmonic approximation, which proves too demanding if performed fully at the given quantum level of theory, are presented and tested. For this purpose, atomic multipoles (up to the quadrupoles) for the amoeba force field are parametrized for all amino acid zwitterions. While the calculated lattice energies of the amino acids range from 235–458 kJ mol−1 in absolute value, the proton transfer energies typically amount to 100–220 kJ mol−1, which translates to sublimation enthalpies ranging from 117–202 kJ mol−1, appreciably exceeding the sublimation enthalpy values common for nonionic molecular crystals. Critically assessed experimental data on sublimation enthalpies are used as a benchmark for comparison of the data calculated in this work. Cohesive properties of most amino acids calculated in this work, combining the PBE-D3(BJ)/PAW and CCSD(T)-F12/aug-cc-pVDZ levels of theory used for predictions of the lattice energies and of the proton transfer energies, respectively, exhibit a reasonable agreement with the experiment. At the same time, this work contains the first published data on cohesive properties for several enantiopure amino acids.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

