
Switching the charge transfer characteristics of quaterthiophene from p-type to n-type via interactions with carbon nanotubes†
 a
and
C. N.
Ramachandran
*a
a
and
C. N.
Ramachandran
*a
Abstract
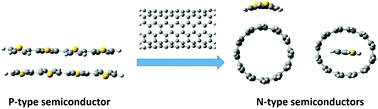
Carbon nanotube-based semiconductors are of great interest for optoelectronic applications at the nanoscale. The present study investigates the structural, optoelectronic and charge transport properties of non-covalent complexes formed between carbon nanotubes (CNTs) and quaterthiophene (4T) by employing dispersion-corrected density functional methods. The effect of different functionals viz., B97-D, B3LYP-GD3 and ωB97X-D on the properties of endo- and exohedral complexes is examined. The endohedral complex (4T@CNT) is found to be energetically more stable than the exohedral one (4T-CNT). Electronic properties such as the ionization energy, electron affinity and energy gap between the frontier molecular orbitals of the CNT are not significantly changed by the adsorption or the encapsulation of 4T. Contrary to the p-type characteristics of 4T, its complexes formed with CNTs exhibit n-type characteristics due to the higher electron mobility than the hole mobility. Among the endo- and exohedral complexes, the former one shows the highest electron mobility of 3.79 cm2 V−1 s−1. The absorption properties of all the systems were studied by time-dependent density functional theory (TD-DFT). It is found that the complexes undergo several charge transfer transitions in the visible region of the electromagnetic spectrum. The above results unequivocally suggest that the charge-transfer characteristics of 4T can be altered on forming complexes with CNTs.
 Please wait while we load your content...
Please wait while we load your content...

