
Triplet state structure–property relationships in a series of platinum acetylides: effect of chromophore length and end cap electronic properties†

Abstract
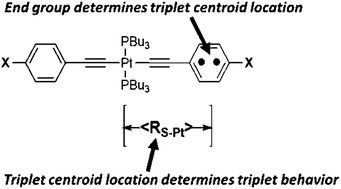
To develop quantitative structure–spectroscopic property relationships in platinum acetylides, we investigated the triplet state behavior of nominally centrosymmetric chromophores trans-Pt(PBu3)2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C-Phenyl-X)2, where X = diphenylamino, NH2, OCH3, t-Bu, CH3, H, F, benzothiazole, CF3, CN, and NO2. We measured ground state absorption, phosphorescence, excitation and triplet state absorption spectra and triplet lifetimes. By DFT we calculated the phosphorescence emission energy (ET), the spin density on the end cap (SD(X)), triplet state geometry and the distance between the triplet centroid and the central platinum atom (〈RS–Pt(X)〉). Compounds with electron-donating X have smaller triplet state lifetime, blue-shifted phosphorescence and larger triplet potential energy surface displacement associated with the CC bond. Compounds with electron-withdrawing X have larger triplet lifetime, red-shifted phosphorescence and smaller triplet potential energy surface displacement associated with the CC bond. The range of spin–orbit-coupling between the platinum atom and the triplet centroid was determined to be 6 Å. The quantity 〈RS–Pt〉 is shown to be a linear function of one-dimensional well length calculated from experimental ET. The multiple examples demonstrate 〈RS–Pt〉 is a useful descriptor for analyzing triplet state behavior.
C-Phenyl-X)2, where X = diphenylamino, NH2, OCH3, t-Bu, CH3, H, F, benzothiazole, CF3, CN, and NO2. We measured ground state absorption, phosphorescence, excitation and triplet state absorption spectra and triplet lifetimes. By DFT we calculated the phosphorescence emission energy (ET), the spin density on the end cap (SD(X)), triplet state geometry and the distance between the triplet centroid and the central platinum atom (〈RS–Pt(X)〉). Compounds with electron-donating X have smaller triplet state lifetime, blue-shifted phosphorescence and larger triplet potential energy surface displacement associated with the CC bond. Compounds with electron-withdrawing X have larger triplet lifetime, red-shifted phosphorescence and smaller triplet potential energy surface displacement associated with the CC bond. The range of spin–orbit-coupling between the platinum atom and the triplet centroid was determined to be 6 Å. The quantity 〈RS–Pt〉 is shown to be a linear function of one-dimensional well length calculated from experimental ET. The multiple examples demonstrate 〈RS–Pt〉 is a useful descriptor for analyzing triplet state behavior.
 Please wait while we load your content...
Please wait while we load your content...

