
Theoretical study of the F(2P) + NH3 → HF + NH2 reaction on an accurate potential energy surface: dynamics and kinetics†

Abstract
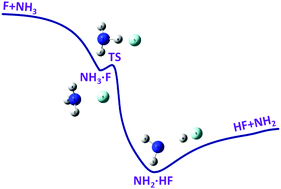
The highly exothermic hydrogen abstraction reaction of the F atom with NH3 is investigated using the quasi-classical trajectory method on a newly developed potential energy surface (PES) for the ground electronic state. The full-dimensional PES is constructed by fitting 41 282 ab initio energy points at the level of UCCSD(T)-F12/aug-cc-pVTZ. The flexible fundamental invariant-neural network method is applied in the fitting, resulting in a total root mean square error of 0.13 kcal mol−1. On one hand, the calculated differential cross sections agree reasonably well with the experimental results and indicate that the reaction is dominated by the direct abstraction and stripping mechanisms while a considerable amount of reaction takes place by the indirect “yo–yo” mechanism. The product energy partition also reproduces well the experimental result, which can be understood according to the geometry change along the minimum energy path. On the other hand, the obtained vibrational state distribution of the product HF follows PνHF=2 ≈ PνHF=1 > PνHF=0 > PνHF=3, less consistent with the scattered experimental results. In addition, the calculated thermal rate coefficients have practically no temperature dependence within the statistical errors.
 Please wait while we load your content...
Please wait while we load your content...

