
Monte-Carlo simulation combined with density functional theory to investigate the equilibrium thermodynamics of electrode materials: lithium titanates as model compounds†

Abstract
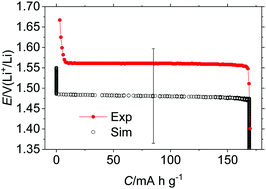
By using lithium titanate (LTO) as a model electrode material, the present study proposes a method to describe its equilibrium thermodynamics based on the Monte-Carlo simulation (MC), for which the energetic parameters are determined by the density functional theory (DFT). The electrochemical potential profile is simulated by a simple topological model which consists only of three parameters representing the Li site energies; namely, the potential energy of the 8a site (ε8a), the difference in the site energy between the 8a and 16c sites (Δε) and the repulsion between two Li atoms situated at the adjacent 8a and 16c sites (J). Parameter physics by the MC revealed that the term Δε plays a decisive role, with a collateral effect from J, for characterizing the shape of the potential profile whereas the term ε8a determines its position along the electrochemical potential. For instance, if Δε exceeds the thermal energy at the temperature under consideration, i.e., if Δε > 3kT, the first-order phase transition takes place during which two phases coexist, resulting in a plateau region in the potential profile. On the other hand, if Δε < 3kT, the lithiation of LTO is viewed as a phenomenon above the critical point, above which the material is in a homogeneous uniphasic state. A multiple regression analysis of a set of the total energy calculated by DFT allows us to determine these energetic terms. The MC simulation with the determined parameters well reproduces the shape and position of the experimental potential profile of LTO. Since the determined value, Δε/eV ∼ 0.4, far exceeds the thermal energy at ambient temperature, the potential plateau of LTO is explained by the first-order phase transition as long as the equilibrium state is concerned.
 Please wait while we load your content...
Please wait while we load your content...

