
Assessing the performance of MM/PBSA and MM/GBSA methods. 9. Prediction reliability of binding affinities and binding poses for protein–peptide complexes†
 a
Tingjun
Hou
*ab
a
Tingjun
Hou
*ab
Abstract
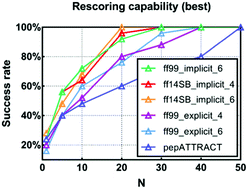
A significant number of protein–protein interactions (PPIs) are mediated through the interactions between proteins and peptide segments, and therefore determination of protein–peptide interactions (PpIs) is critical to gain an in-depth understanding of the PPI network and even design peptides or small molecules capable of modulating PPIs. Computational approaches, especially molecular docking, provide an efficient way to model PpIs, and a reliable scoring function that can recognize the correct binding conformations for protein–peptide complexes is one of the most important components in protein–peptide docking. The end-point binding free energy calculation methods, such as MM/GBSA and MM/PBSA, are theoretically more rigorous than most empirical and semi-empirical scoring functions designed for protein–peptide docking, but their performance in predicting binding affinities and binding poses for protein–peptide systems has not been systematically assessed. In this study, we first evaluated the capability of MM/GBSA and MM/PBSA with different solvation models, interior dielectric constants (εin) and force fields to predict the binding affinities for 53 protein–peptide complexes. For the 19 short peptides with 5–12 residues, MM/PBSA based on the minimized structures in explicit solvent with the ff99 force field and εin = 2 yields the best correlation between the predicted binding affinities and the experimental data (rp = 0.748), while for the 34 medium-size peptides with 20–25 residues, MM/GBSA based on 1 ns of molecular dynamics (MD) simulations in implicit solvent with the ff03 force field, the GBOBC1 model and a low interior dielectric constant (εin = 1) yields the best accuracy (rp = 0.735). Then, we assessed the rescoring capability of MM/PBSA and MM/GBSA to distinguish the correct binding conformations from the decoys for 112 protein–peptide systems. The results illustrate that MM/PBSA based on the minimized structures with the ff99 or ff14SB force field and MM/GBSA based on the minimized structures with the ff03 force field show excellent capability to recognize the near-native binding poses for the short and medium-size peptides, respectively, and they outperform the predictions given by two popular protein–peptide docking algorithms (pepATTRACT and HPEPDOCK). Therefore, MM/PBSA and MM/GBSA are powerful tools to predict the binding affinities and identify the correct binding poses for protein–peptide systems.
 Please wait while we load your content...
Please wait while we load your content...

