
Global and local aromaticity of acenes from the information-theoretic approach in density functional reactivity theory

Abstract
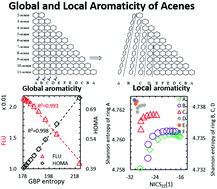
In this work, we report a systematic study on the global and local aromaticity of acenes using a series of model structures from 2-acene to 11-acene. A recently developed ansatz, an information-theoretic approach coached into density functional reactivity theory has been employed, which essentially provides different density functionals characterizing the molecular electron density distribution. Based on the correlation analysis of six conventional aromaticity indices with eight information-theoretic quantities, we examined the aromaticity of acenes from both global and local perspectives. From the global aromaticity viewpoint, our results suggest that different descriptors based on various physicochemical properties are intrinsically dependent. A novel laminated feature ruling local aromaticity of acenes has been unveiled, from which we found that the distance from the terminal rings plays the critical role. Based on the shape of the correlation plots between the conventional aromaticity indices and information-theoretic quantities, the latter could be separated into three subgroups. The seemingly contradictory results from global and local aromaticity perspectives not only present us the uniqueness of the acene systems but all demonstrate the effectiveness of the information-theoretic approach from density functional reactivity theory. Besides strengthening the validity of a series of new aromaticity descriptors, our results should lead to more clear insights into the chemical significance of the information-theoretic quantities.
 Please wait while we load your content...
Please wait while we load your content...

