
Actinyl cation–cation interactions in the gas phase: an accurate thermochemical study†

Abstract
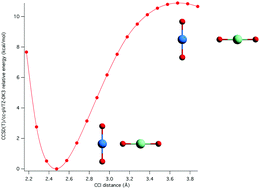
Gas phase actinyl cation–cation interactions (CCIs) were studied by an accurate composite coupled cluster thermochemical approach for the first time. A number of CCI dimers were constructed from the monomers UO22+, UO2+, NpO22+, NpO2+, PuO2+, and AmO2+. All CCI dimers studied were calculated to be thermodynamically unstable, with dissociation energies ranging from −60 to −90 kcal mol−1, but in many cases kinetic stability was indicated by calculated local minima with well depths as large as ∼15 kcal mol−1. Most of the dimers studied involved a T-shaped geometry, although one side-on dimer, (UO2+)2, was included since it was amenable to coupled cluster methods. In the T-shaped isomers the most stable dimers were calculated to arise when the oxo-group of an An(V) actinyl cation was oriented towards the metal center of an An(VI) actinyl cation. For both mixed-valent An(VI)/An(V) and mono-valent An(V) dimers, the stability as estimated from the depth of the calculated local minimum decreased in the donor series U(V) > Np(V) > Pu(V) > Am(V). These trends correlate well with experimental trends in condensed phase CCIs. A rationale for the bonding in CCIs was investigated by carrying out charge transfer analyses using the natural bond orbital (NBO) method. Augmenting the usual Lewis acid–base explanation, CCIs are the direct result of a competition between charge transfer stabilization, which can be as much as 0.11e or 30.7 kcal mol−1 at equilibrium, and Coulombic repulsive destabilization.
 Please wait while we load your content...
Please wait while we load your content...

