
Absorption shifts of diastereotopically ligated chlorophyll dimers of photosystem I†
 a
Heike
Fliegl,
b
Evgeni B.
Starikov,
c
Ville R. I.
Kaila
e
and
Dage
Sundholm
*fg
a
Heike
Fliegl,
b
Evgeni B.
Starikov,
c
Ville R. I.
Kaila
e
and
Dage
Sundholm
*fg
Abstract
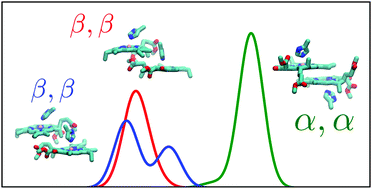
The light-harvesting chlorophyll (Chl) molecules of photosynthetic systems form the basis for light-driven energy conversion. In biological environments, the Chl chromophores occur in two distinct diastereotopic configurations, where the α and β configurations have a magnesium-ligating histidine residue and a 17-propionic acid moiety on the opposite side or on the same side of the Chl ring, respectively. Although β-ligated Chl dimers occupy conserved positions around the reaction center of photosystem I (PSI), the functional relevance of the α/β configuration of the ligation is poorly understood. We employ here correlated ab initio calculations using the algebraic-diagrammatic construction through second order (ADC(2)) and the approximate second-order coupled cluster (CC2) methods in combination with the reduced virtual space (RVS) approach in studies of the intrinsic excited-state properties of α-ligated and β-ligated Chl dimers of PSI. Our ab initio calculations suggest that the absorption of the α-ligated reaction-center Chl dimer of PSI is redshifted by 0.13–0.14 eV in comparison to the β-ligated dimers due to combined excitonic coupling and strain effects. We also show that time-dependent density functional theory (TDDFT) calculations using range-separated density functionals underestimate the absorption shift between the α- and β-ligated dimers. Our findings may provide a molecular starting point for understanding the energy flow in natural photosynthetic systems, as well as a blueprint for developing new molecules that convert sunlight into other forms of energy.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

