
Phenylacetylene as a gas phase sliding balance for solvating alcohols†

Abstract
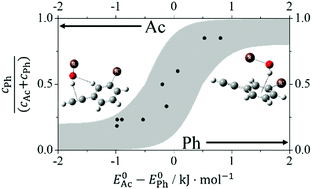
Phenylacetylene offers two similarly attractive π binding sites to OH containing solvent molecules, the phenyl ring and the acetylenic triple bond. By systematically varying the solvent molecule and by methylating aromatic or acetylenic CH groups, the docking preference can be controlled. It ranges from almost exclusive acetylene docking to predominant phenyl docking, depending on how electron density is deposited into the conjugated system and how large the London dispersion interaction is. FTIR spectroscopy of supersonic jet expansions is used to observe the competitive docking preferences in phenylacetylene and some of its methylated derivatives. A new data evaluation procedure that estimates band strength uncertainties based on a Monte Carlo approach is introduced. We test how well two density functionals (B3LYP-D3 and M06-2X) in combination with a def2-TZVP basis set are able to describe the docking switch. B3LYP-D3 is slightly biased towards acetylenic hydrogen bond docking and M06-2X is strongly biased towards phenyl hydrogen bond docking. More accurate theoretical predictions are invited and some previous experimental assignments are questioned.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

