
First-principles calculations of iodine-related point defects in CsPbI3

Abstract
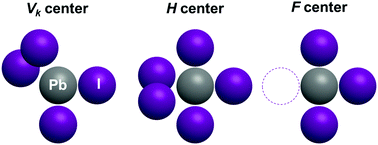
We present here first principles hybrid functional calculations of the atomic and electronic structure of several iodine-related point defects in CsPbI3, a material relevant for photovoltaic applications. We show that the presence of neutral interstitial I atoms or electron holes leads to the formation of di-halide dumbbells of I2− (analogous to the well-known situation in alkali halides). Their formation and one-electron energies in the band gap are determined. The formation energy of the Frenkel defect pair (I vacancies and neutral interstitial I atoms) is found to be ∼1 eV, and as such is smaller than the band gap. We conclude that both iodine dumbbells and iodine vacancies could be, in principle, easily produced by interband optical excitation.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

