
Excited state properties of a series of molecular photocatalysts investigated by time dependent density functional theory†

Abstract
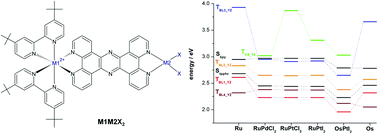
Time dependent density functional theory calculations are applied on a series of molecular photocatalysts of the type [(tbbpy)2M1(tpphz)M2X2]2+ (M1 = Ru, Os; M2 = Pd, Pt; X = Cl, I) in order to provide information concerning the photochemistry occurring upon excitation of the compounds in the visible region. To this aim, the energies, oscillator strengths and orbital characters of the singlet and triplet excited states are investigated. The structural modifications of the complexes have a strong impact on the excited states properties. In particular, it is found that the main differences concern the energies of the charge-separated and metal-centered states. The analysis of these differences provides general trends for the efficiency of population transfers between the states, particularly regarding the charge separation and electron recombination processes.
- This article is part of the themed collection: Bunsentagung 2019: Functional Materials
 Please wait while we load your content...
Please wait while we load your content...

