
First principles calculations of surface dependent electronic structures: a study on β-FeOOH and γ-FeOOH†

Abstract
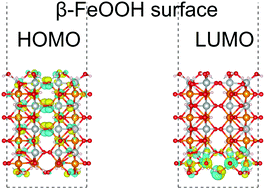
We report a theoretical study on iron oxyhydroxide (FeOOH). The FeOOH surface is expected to act as an efficient electrochemical catalyst for the oxygen evolution reaction (OER), because it is based on iron, an element of the fourth highest Clarke number. Experimentally, the OER activity of β-FeOOH is known to be higher than that of γ-FeOOH. However, the details of the OER mechanism and the surface reactivities of the FeOOH polymorphs have not yet been fully understood. We performed first-principles calculations of bulk and surfaces of β-FeOOH and γ-FeOOH using density functional theory, to investigate their electronic structures and catalytic activities. The calculations suggest that depending on the surface indices, several surfaces may be favored for catalytic activities.
 Please wait while we load your content...
Please wait while we load your content...

