
Double-well potential energy surface in the interaction between h-BN and Ni(111)†

Abstract
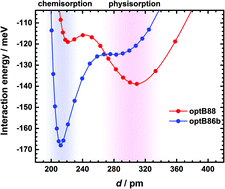
Density functional theory calculations with non-local correlation functionals, properly accounting for dispersion forces, predict the presence of two minima in the interaction energy between h-BN and Ni(111). These can be described as a physisorbed state with no corrugation of the h-BN structure, and a chemisorbed state exhibiting noticeable corrugation and a shorter distance of h-BN to the metallic support. The latter corresponds indeed to the one reported in most experiments. The relative stability of the two minima depends on the specific density functional employed: of those investigated here only optB86b-vdW yields the correct order of stability. We also demonstrate that the effect of the metal support on the Raman frequency of the chemisorbed boron nitride monolayer cannot be reduced to the associated strain. This is important because the Raman frequency has been proposed as a signature to identify h-BN monolayers from multilayered samples. Our analysis shows that such signatures would be strongly dependent on the nature of the interaction between the support and h-BN.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

