
The effect of two types of dibenzoannulation of pentalene on molecular energies and magnetically induced currents†
 *a
and
*a
and
Abstract
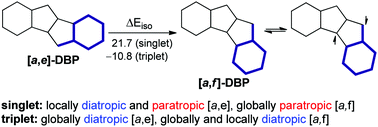
The effect of two types of dibenzo-fusion of pentalene in the singlet and triplet states on its molecular energies and magnetically induced ring currents was examined via density functional calculations. The isomerization energy decomposition analysis (IEDA) together with the calculated aromaticity indices (NICS(1)zz, HOMA and FLUπ), estimation of resonance energies (RE) and extra cyclic resonance energies (ECRE) via the NBO method, and the NICS-XY-scans revealed that the π-electronic system is the most important factor controlling the molecular energies. The [a,f] topology features greater delocalization, which results in two opposing effects: larger ECRE, but weaker π-bonding. The latter is mainly responsible for the higher energy of [a,f]-dibenzopentalene (DBP) (ΔEiso = 21.7 kcal mol−1), with the other effects being σ-orbital and electrostatic interactions. The reversal of energetic stability in the triplet states (ΔEiso = −10.8 kcal mol−1) mainly comes from the reduced Pauli repulsion in [a,f]-DBP, which stabilizes the unpaired spin density over the central trimethylenemethane subunit vs. the central pentalene subunit in [a,e]-DBP. Although the [a,e] topology only reduces the diatropic and paratropic currents of the elementary subunits, benzene and pentalene, the [a,f] topology also creates strong global paratropicity involving the benzene rings. Both DBP isomers are characterized by global and smaller semi-global and local diatropic currents in the triplet state.
 Please wait while we load your content...
Please wait while we load your content...

