
A variationally computed room temperature line list for AsH3†

Abstract
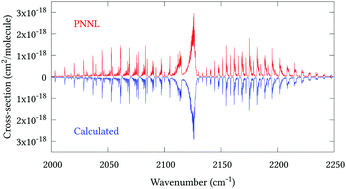
Calculations are reported on the rotation–vibration energy levels of the arsine molecule with associated transition intensities. A potential energy surface (PES) obtained from ab initio electronic structure calculations is refined to experimental data, and the resulting energy levels display sub-wavenumber accuracy for all reliably known J = 0 term values under 6500 cm−1. After a small empirical adjustment of the band centres, our calculated (J = 1–6) rovibrational states reproduce 578 experimentally derived energies with a root-mean-square error of 0.122 cm−1. Absolute line intensities are computed using the refined PES and a new dipole moment surface (DMS) for transitions between states with energies up to 10 500 cm−1 and rotational quantum number J = 30. The computed DMS reproduces experimental line intensities to within 10% uncertainty for the ν1 and ν3 bands. Furthermore, our calculated absorption cross-sections display good agreement with the main absorption features recorded in the Pacific Northwest National Laboratory (PNNL) for the complete range of 600–6500 cm−1.
 Please wait while we load your content...
Please wait while we load your content...

