
Can microsolvation effects be estimated from vacuum computations? A case-study of alcohol decomposition at the H2O/Pt(111) interface†
 a
and
Carine
Michel
*a
a
and
Carine
Michel
*a
Abstract
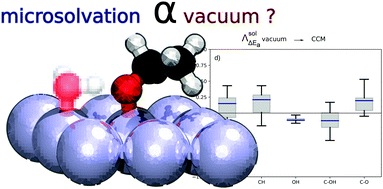
Converting biomass into sustainable chemicals and energy feed-stocks requires innovative heterogeneous catalysts, which are able to efficiently work under aqueous conditions. Computational chemistry is a key asset in the design of these novel catalysts, but it has to face two challenges: the large reaction networks and the potential role of hydration. They can be addressed using scaling relations such as Brønsted–Evans–Polanyi (BEP) and solvation models, respectively. In this study, we show that typical reaction and activation energies of alcohol decomposition on Pt(111) are not strongly modified by the inclusion of the water solvent as a continuum model. In contrast, adding a single water molecule strongly favours O–H and C–OH scission and it prevents C–O and to a lesser extent C–C scissions. The resulting BEP relationships partially reflect these changes induced by the solvent. Predicting Pt-catalysed alcohol decomposition in water should thus account for the influence of the solvent on thermodynamics and kinetics. In addition, we found that the reaction energies obtained in the presence of an explicit water molecule scale with the ones obtained in a vacuum. Hence, we reveal that vacuum computations in combination with corrections based on our linear regressions are able to capture the important H-bonding effect.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

