
The dynamical interplay between a megadalton peptide nanocage and solutes probed by microsecond atomistic MD; implications for design†

Abstract
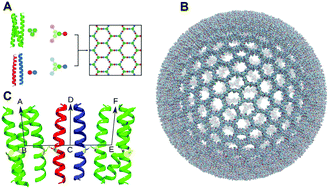
Understanding the assembly and dynamics of protein-based supramolecular capsids and cages is of fundamental importance and could lead to applications in synthetic biology and biotechnology. Here we present long and large atomistic molecular dynamics simulations of de novo designed self-assembling protein nanocages (SAGEs) in aqueous media. Microsecond simulations, comprised of ≈42 million atoms for three pre-formed SAGEs of different charges, in the presence of solutes and solvent have been completed. Here, the dynamics, stability and porosity of the peptide networks are explored along with their interactions with ions, small molecules and macromolecular solutes. All assemblies are stable over the μs timescale, and the solutes show a mixture of transport behaviour across or adherence to the fabric of the SAGE particles. Solute proteins largely retained native-like conformation on contact with SAGE. Certain residues of the SAGE peptides are identified as “repeat offenders” for contacting many different solutes, which suggest modifications to reduce non-specific binding. These studies highlight how molecular dynamics can aid the design process of SAGE and similar assemblies for potential applications as diverse as platforms for drug and vaccine delivery and nanoreactors to encapsulate enzyme pathways.
 Please wait while we load your content...
Please wait while we load your content...

