
Theoretical investigation of the isomerization pathways of diazenes: torsion vs. inversion†

Abstract
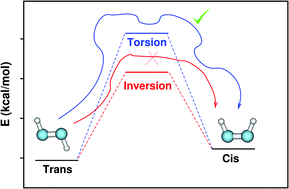
Diazenes are an important family of organic compounds used widely in synthetic and materials chemistry. These molecules have a planar geometry and exhibit cis–trans isomerization. The simplest of all these molecules – diazene (N2H2) – has been subjected to several experimental and theoretical studies. Two mechanisms have been proposed for the cis–trans isomerization of diazene, which are an in-plane inversion and an out-of-plane torsion. The activation energies for these pathways are similar and the competition between these two mechanisms has been discussed in the literature based on electronic structure theory calculations. Three decades ago, a classical dynamics investigation of diazene isomerization was carried out using a model Hamiltonian and it was indicated that the in-plane inversion is forbidden classically because of a centrifugal barrier and the out-of-plane torsion is the only isomerization pathway. In the present work, we investigated the cis–trans isomerization dynamics of diazene using ab initio classical trajectory simulations at the CASSCF(2,2)/aug-cc-pVDZ level of electronic structure theory. The simulation results confirmed the presence of the aforementioned centrifugal barrier for the inversion and torsion was the only observed pathway. The calculations were repeated for a similar system (difluorodiazene, N2F2) and again the centrifugal barrier prevented the inversion pathway.
 Please wait while we load your content...
Please wait while we load your content...

