
Understanding water interactions with organic surfaces: environmental molecular beam and molecular dynamics studies of the water–butanol system
 a
Josip
Lovrić,
ab
Xiangrui
Kong,
a
Erik S.
Thomson,
a
Céline
Toubin
b
and
Jan B. C.
Pettersson
*a
a
Josip
Lovrić,
ab
Xiangrui
Kong,
a
Erik S.
Thomson,
a
Céline
Toubin
b
and
Jan B. C.
Pettersson
*a
Abstract
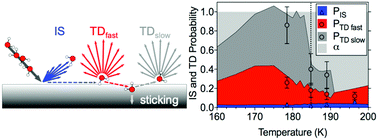
The interactions between water molecules and condensed n-butanol surfaces are investigated at temperatures from 160 to 240 K using the environmental molecular beam experimental method and complementary molecular dynamics (MD) simulations. In the experiments hyperthermal water molecules are directed onto a condensed n-butanol layer and the flux from the surface is detected in different directions. A small fraction of the water molecules scatters inelastically from the surface while losing 60–90% of their initial kinetic energy in collisions, and the angular distributions of these molecules are broad for both solid and liquid surfaces. The majority of the impinging water molecules are thermalized and trapped on the surface, while subsequent desorption is governed by two different processes: one where molecules bind briefly to the surface (residence time τ < 10 μs), and another where the molecules trap for a longer time τ = 0.8–2.0 ms before desorbing. Water molecules trapped on a liquid n-butanol surface are substantially less likely to escape from the surface compared to a solid layer. The MD calculations provide detialed insight into surface melting, adsorption, absorption and desorption processes. Calculated angular distributions and kinetic energy of emitted water molecules agree well with the experimental data. In spite of its hydrophobic tail and enhanced surface organization below the melting temperature, butanol's hydrophilic functional groups are concluded to be surprisingly accessible to adsorbed water molecules; a finding that may be explained by rapid diffusion of water away from hydrophobic surface structures towards more strongly bound conformational structures.
- This article is part of the themed collections: 2019 PCCP HOT Articles and 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

