
Stacking interactions between ruthenium p-cymene complexes: combined crystallographic and density functional study†
 a
and
Snežana D.
Zarić
*ab
a
and
Snežana D.
Zarić
*ab
Abstract
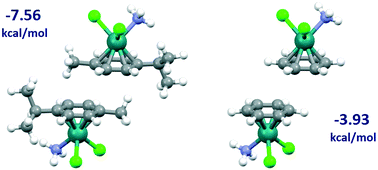
A search of the Cambridge Structural Database for stacking interactions between p-cymene (1-methyl-4-isopropylbenzene) ligands of transition metal complexes revealed three preferred interaction geometries, all with an antiparallel orientation. The most frequent one involves both stacking of aromatic rings and C–H/π interactions of methyl substituents with aromatic rings, while the second most frequent has stacking of aromatic rings and C–H/π interactions of methyl groups of isopropyl substituents with aromatic rings. The results of the CSD search are in agreement with the DFT calculations of interaction energies, since all the preferred CSD geometries correspond to minima on potential energy curves. The strongest calculated interaction between p-cymene ligands of model complex [Ru(p-cym)Cl2(NH3)], with a B97-D2/def2-TZVP interaction energy of −7.56 kcal mol−1, corresponds to the most frequent geometry found in crystal structures, that contain mostly ruthenium complexes. This is significantly stronger than the interaction between benzene ligands of [Ru(benzene)Cl2(NH3)] complexes (−3.93 kcal mol−1), revealing that substituents increase their interaction strength substantially. All interaction geometries and their relative strengths are in agreement with the electrostatic potential of the monomer complex.
 Please wait while we load your content...
Please wait while we load your content...

