
Molecular dynamics simulation of homogeneous nucleation of supersaturated potassium chloride (KCl) in aqueous solutions†

Abstract
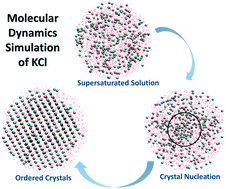
Molecular dynamics (MD) simulation is used to investigate the mechanism of crystal nucleation of potassium chloride (KCl) in a supersaturated aqueous solution at 293 K and 1 atm. Using radial distribution function (RDF), bond distance calculation, angle measurement, and configurational snapshots, it was found that the newly emerged phase is face-centered cubic crystals with some water molecules trapped inside the crystal lattice. It was also shown that during early stages of nucleation, high local density of ions occurred, and within these areas, the nucleation started with a sequence of ionic additions, as suggested by classical nucleation theory. It was concluded that crystal nuclei form in a sequential manner, but this can only happen in places where the local density of ions (supersaturation) is higher than the solution concentration, and the probability of having effective collisions increases, making these sites primary candidates for nucleation.
 Please wait while we load your content...
Please wait while we load your content...

