
An XRD and NMR crystallographic investigation of the structure of 2,6-lutidinium hydrogen fumarate†

Abstract
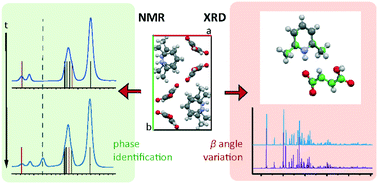
Fumarate is a pharmaceutically acceptable counterion often used to modify the biophysical properties of active pharmaceutical ingredients (APIs) through salt formation. With 2,6-lutidine (2,6-dimethylpyridine), fumaric acid forms the salt 2,6-lutidinium hydrogen fumarate. An NMR crystallography approach was employed to investigate the salt structure and the intermolecular interactions involved in its formation and stability. The crystallographic unit cell was determined by both single crystal XRD (SXRD) and synchrotron powder X-ray diffraction (PXRD) to contract at low temperature with a skew in the β angle. Density functional theory (DFT)-based geometry optimisations were found partially to replicate this. A second room temperature structure was also identified which exhibited a similar skew of the β angle as the low temperature structure. DFT calculation was also employed, alongside 2D 1H double-quantum (DQ) magic angle spinning (MAS) and 14N–1H HMQC MAS NMR spectra, to investigate the hydrogen bonding network involved in the structure. DFT-based gauge-including projector-augmented wave (GIPAW) calculations highlighted both strong N+–H⋯O− and O–H⋯O intermolecular hydrogen bonds between the molecules, as well as several weaker CH⋯O hydrogen bonds. Both PXRD and solid-state MAS NMR, supported by thermal gravimetric analysis (TGA) and solution-state NMR analysis, show formation of fumaric acid within samples over time. This was evidenced by the identification of reflections and peaks associated with crystalline fumaric acid in the PXRD pattern and in 1H MAS and 13C cross polarization (CP) MAS solid-state NMR spectra, respectively.
 Please wait while we load your content...
Please wait while we load your content...

