
Selective CO2 adsorption and theoretical simulation of a stable Co(ii)-based metal–organic framework with tunable crystal size†

Abstract
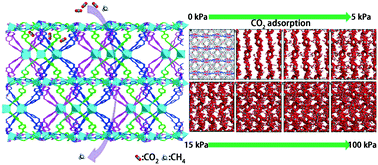
A three-fold interpenetrated metal–organic framework [Co2(OBA)4(PTD)·3DMF·CH3CH2OH·5H2O]n (1) has been synthesized by utilizing 4,4′-oxybis(benzoic acid) (H2OBA) as the linker, 6-(pyridin-4-yl)-1,3,5-triazine-2,4-diamine (PTD) as the ligand, and CoCl2·6H2O via solvothermal method. Compound 1 exhibits not only a high uptake capacity for CO2 molecules with an estimated high sorption heat (50.6 kJ mol−1 at zero loading), but also a significant selective adsorption of CO2 over CH4, which may be ascribed to the presence of proper-sized pores with high polarity, amine groups and triazine rings of PTD linker decorating the pores. Meanwhile, the Grand Canonical Monte Carlo (GCMC) simulations of CO2 adsorption of compound 1 demonstrate that CO2 molecules are preferentially adsorbed around the PTD ligands. Furthermore, complex 1 displays a relatively high adsorption capacity of H2 (101.7 cm3 g−1 at 1 bar) under 77 K.
 Please wait while we load your content...
Please wait while we load your content...

