
Increasing topological diversity during computational “synthesis” of porous crystals: how and why†

Abstract
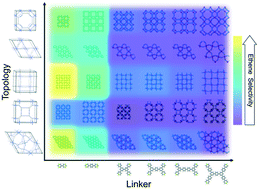
Effectively tuning the properties of porous crystals could lead to breakthroughs in areas such as molecular separation, chemical sensing, and catalysis. The most tunable kind of porous crystals are metal–organic frameworks (MOFs). Their tunability has resulted in MOFs being the focus of intensive research. However, MOF tunability also gives rise to an overwhelmingly large “MOF-space” (the conceptual space of all MOFs) that cannot be comprehensively explored experimentally. Because of this, high throughput computational screening (HTCS) emerged as a tool to more rapidly explore MOF-space. However, the effectiveness of HTCS is tied to the ability of computational algorithms for automated crystal construction to access every point in MOF-space. To this end, here we introduce a crystallographic net rescaling algorithm to the topologically-based crystal constructor (ToBaCCo) code that allows for the automated construction of MOFs (or other porous crystals) of any topology by exploiting graph theory. We demonstrate the capabilities of the new version of the code (ToBaCCo 3.0) by computationally “synthesizing” seven sets of isomorphic MOFs, where chemical composition remains constant within each set, but crystal topology varies across 19 previously inaccessible topologies. Furthermore, we illustrate the importance of computationally synthesizing all possible isomorphs for a given MOF composition by showing how strongly crystal topology influences simulated adsorption and mechanical properties. Specifically, we show that among crystal isomorphs i) methane deliverable capacity can vary by values as large as 150 cc(STP)/cc ii) adsorption selectivity for ethene/ethane mixtures can switch from ethene- to ethane-selective between MOF isomorphs, and iii) mechanical stability can be lost, and bulk moduli can exhibit significant variations, all solely due to a change in crystal topology.
 Please wait while we load your content...
Please wait while we load your content...

